

 06 Jun
06 Jun 
-

+86 18962587269
-

contact@readcrystal.com
-

Changshu High-tech Industrial Development Zone, Suzhou, Jiangsu Province

With the increasing understanding of the three-dimensional structure of drug target proteins and the deepening understanding of the interaction sites between target proteins and lead compounds or natural ligands, Structure-Based Drug Design (SBDD) has emerged. Rapid advances in protein expression, purification, and protein crystallography have provided detailed structural information for disease-relevant protein targets. Simultaneously, the evolution of chemical synthesis aligns perfectly. New effective reagents, protective groups, catalytic transfer, and multi-step synthesis strategies provide huge innovation potential for structure-based drug design. The method of drug design based on structure has revolutionized the research strategy of medicinal chemistry and changed the methods of screening and optimization of new drugs.
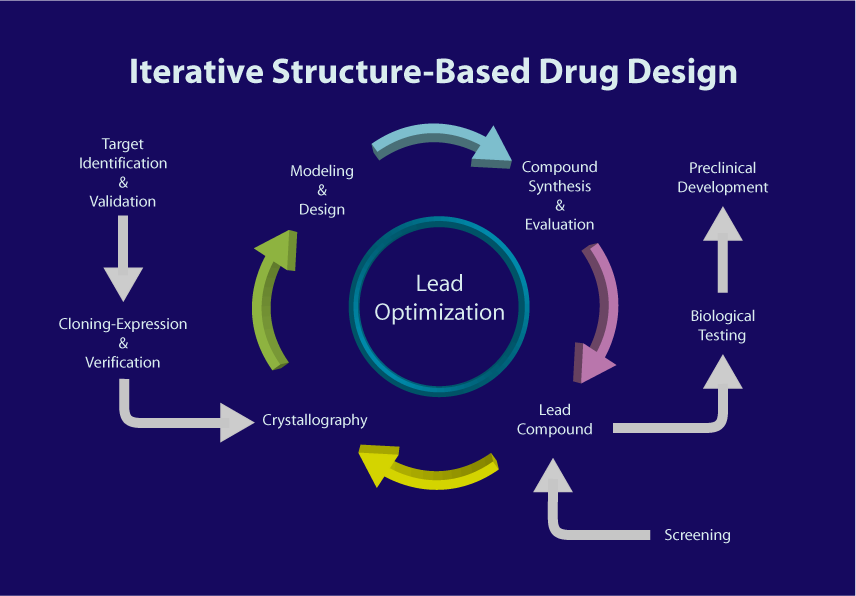
Figure 1. Structure-based drug design process
Roche has used the S BDD method to develop a protease inhibitor Saquinavir. This drug makes the treatment of HIV-AIDS possible and shows the great potential of this design method. Subsequently, many new drugs were developed to treat hypertension, HIV/AIDS, various cancers, and other human diseases. Recently, SBDD has been used in the development of drugs to treat COVID-19.
The structure-based design strategy requires first obtaining information on the shape and charge characteristics of the target protein binding site. Then measuring and analyzing the crystal structure of the protein-ligand complex to obtain information on the interaction between the two. This molecular-level structural analysis frequently provides active conformations of ligands essential for molecular design. This critical information fuels structure-based design strategies, allowing for the optimization of the structure while maintaining its framework of the lead compound. Consequently, this process improves the potency and selectivity of the drug. An important discipline in this process is macromolecular crystallography.
In the following sections, we will introduce the role of macromolecular crystallography at every stage of SBDD, and its guidance in the development of innovative drugs.
A.Target identification and selection
1.Protein function prediction
In modern drug research and development, the initial stage is to identify, determine, and prepare drug screening targets - molecular drug targets. Statistical analysis shows that over 50% of existing drugs in the market target protein receptors, which reflects the importance of proteins in this field. The function of a protein is mainly verified through experiments. In instances where experiments fail to yield the required information, it can also be derived from the sequence similarity of the protein. However, a large number of protein sequences lack close similarity to known functional proteins. In such scenarios, predicting and analyzing the protein structure becomes essential to obtain functional clues.
Predicting protein function from its structure relies significantly on the following approaches:
1) Folding Patterns
Proteins with similar functions usually have similar folding patterns. Finding proteins with similar folding directions in structure through alignment and matching analysis is the first step in structure-based function prediction.
2) Surface clefts and binding pockets
The clefts (tube-like, groove-like, shallow depression-like areas) and pockets (pocket-like areas) on the protein surface are also important clues for inferring its function. These regions often serve as interaction sites for other proteins, substrates, and regulatory elements. For example, enzymes commonly have significantly functional sites within the largest clefts on their surfaces, with one of these often representing the active or catalytic site.
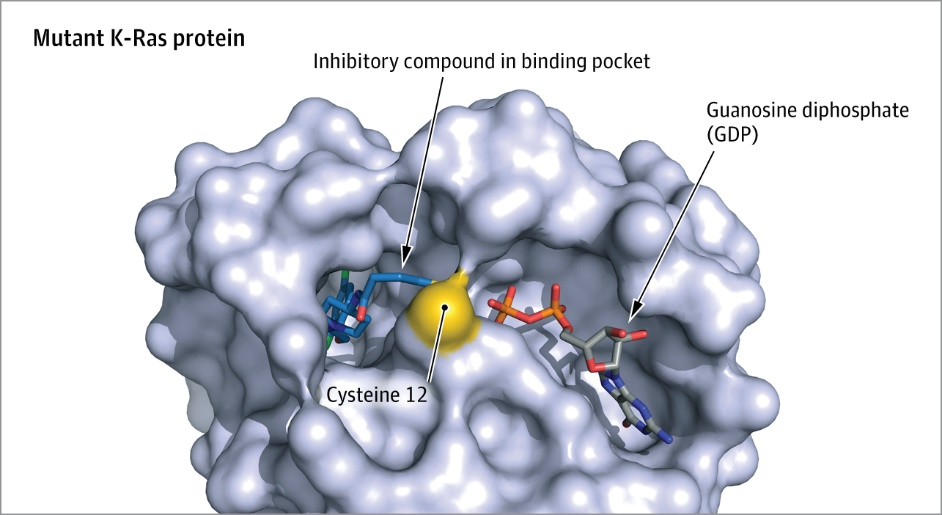
Figure 2. Schematic diagram of the drug binding pocket structure in K-Ras protein
3) Residue Analysis
Proteins with a specific function often have evidence of being affected by a few residues in a confined region of their three-dimensional structure. Whether the catalytic residues in enzyme active site or specific residues in DNA-binding protein crucial for binding certain DNA sequences, the arrangement and conformation of residues are critical to the function of the protein and are highly conserved throughout evolution.
4) Phylogenetic Relationships
Essential regions in protein structures determine function and remain highly conserved during evolution. Analysis of phylogenetic Relationships or identifying determinant residues within evolutionary trees aids in evaluating protein function.
5) Machine Learning Applications
Utilizing machine learning techniques enables the inference of protein function based on structural similarities. This includes predicting catalytic residues, protein-protein binding sites, and classifying enzymes within families.
Druggability evaluation
The evaluation effectiveness of a drug relies heavily on its ability to bind effectively to a specific pocket on the intended protein target. These binding pockets fall into two key categories: one facilitates the access of a drug to the site of action, while the other enables the crucial interaction between the drug and the molecular target once it reaches this site.

Figure 3. Judgment of druggability of molecules based on protein structure.
The general criteria for determining a druggable target are the presence of a binding site of optimal dimensions (typically capable of accommodating compounds with a molecular weight of around 500 Da), favorable lipophilicity, and sufficient hydrogen bonding sites. These properties can be assessed from the protein crystal structure.
In addition, with the presence of allosteric binding sites and different structural conformations in the protein, the druggable binding can be greatly increased.
Evaluating the potential target of a drug mainly involves predicting whether the protein possesses a binding site that complements the structure of the drug. There are two main methods used to automatically identify ligand binding sites: The first method is geometric analysis, observing the 2D or 3D protein conformation to identify a suitable binding pocket. Proteins can be divided into three regions in terms of spatial structure: the core body, an internal dense region composed of the protein atoms; the solvent contact interface, the outer surface where the protein contacts with the surrounding solvent; and the cavity, which extends from the solvent contact interface into a recessed space in the protein main body. The drug-binding pocket is generally within this cavity region. In addition, evaluation can be based on the specific physical and chemical properties of the protein cavity surface. A druggable protein target should have physicochemical properties that mirror those of the drug-like molecule itself. Therefore, assessing the size, shape characteristics, and measurement of hydrophobicity (i.e., the ratio of hydrogen bond donors and acceptors, and the distribution of polar and hydrophobic atoms within the protein cavity) are important evaluations when identifying drug binding pockets.
To evaluate the druggability of a target based on its structure, structural algorithms are first utilized to predict and assess potential drug-binding sites. Second, specific discriminant functions based on the physicochemical properties of the protein binding pocket are applied. Third, a reference set of targets with known established druggability outcomes serves as a benchmark to screen and compare and screen a large compound library.
+86 18962587269
contact@readcrystal.com
Changshu High-tech Industrial Development Zone, Suzhou, Jiangsu Province

